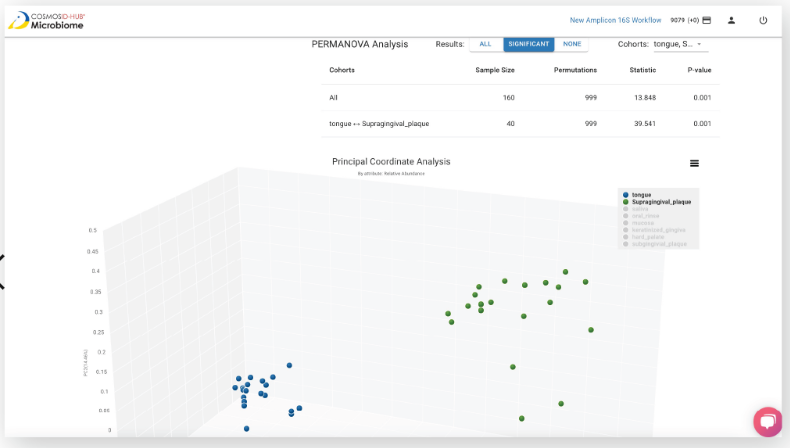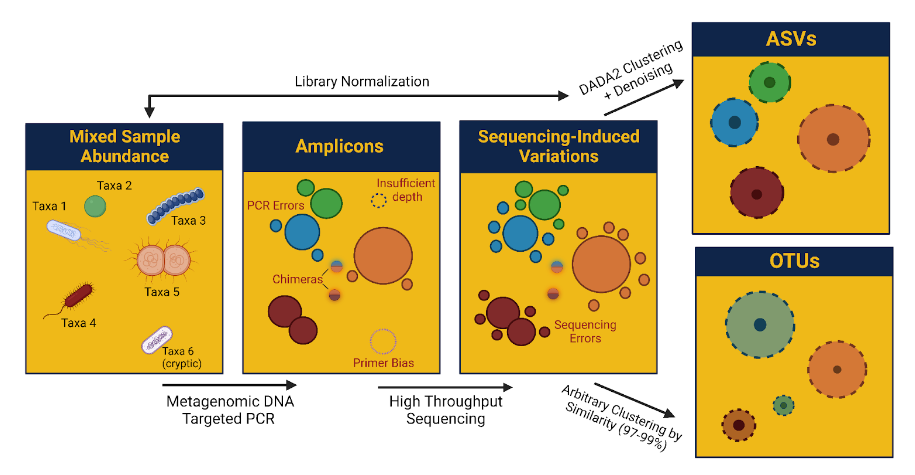
"We've been using CosmosID-HUB for our analysis needs and it allows for detailed analysis and has allowed us to answer important clinical questions."

Jessica Allegretti
Director, Crohn's & Colitis Center & FMT Program at Brigham & Women's Hospital